医疗器械律师注册申报服务
- 网络
- 刘伟律师
- 2017-11-11
医疗器械注册申报服务
1.法规依据
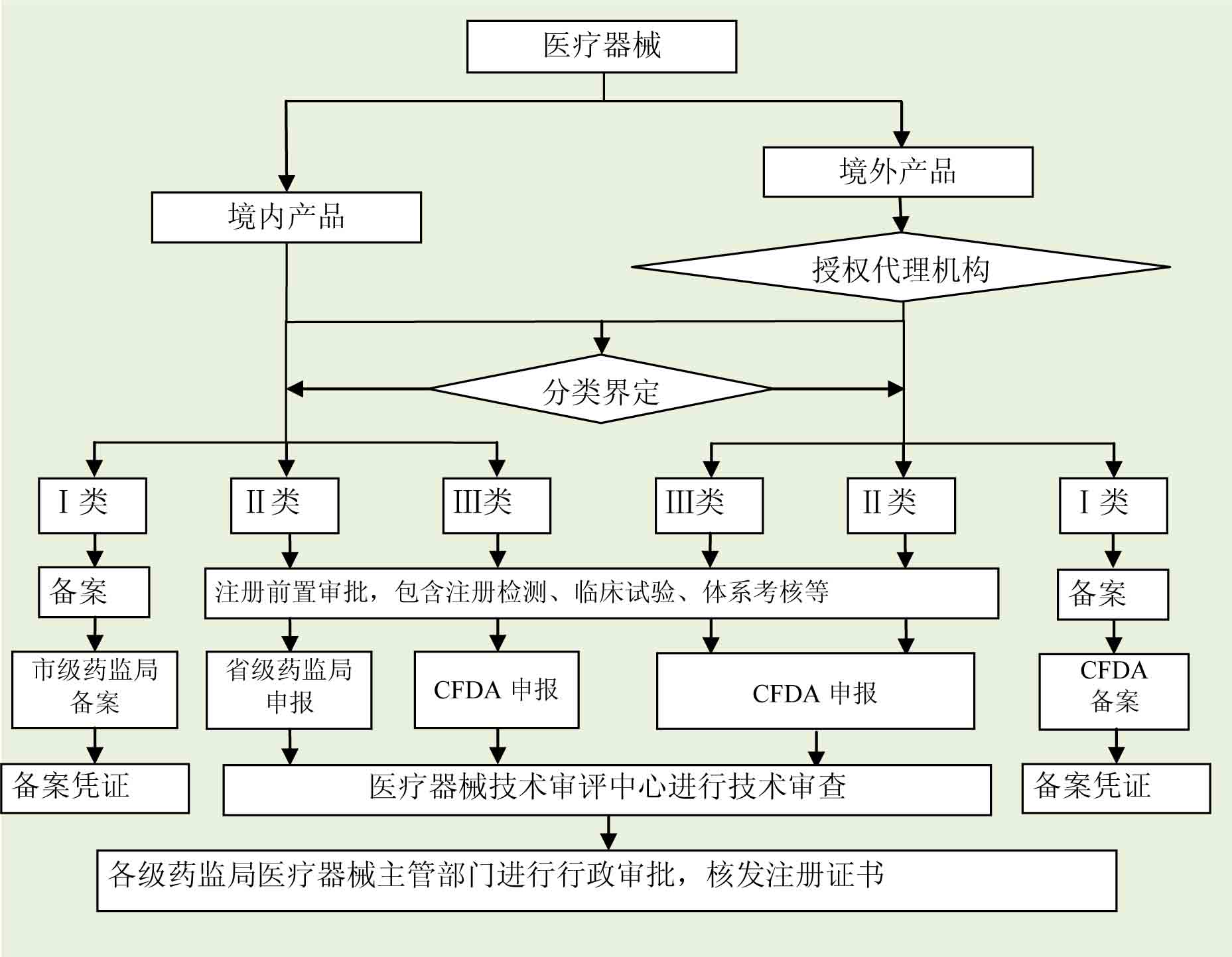
根据《医疗器械监督管理条例》(国务院令第650号)的规定,对医疗器械按照风险程度实行分类管理。
第Ⅰ类是风险程度低,实行常规管理可以保证其安全、有效的医疗器械,境内生产企业需要在所在地市药监局进行备案,境外医疗器械生产企业需要委托境内代理人在CFDA进行备案。
第Ⅱ类是具有中度风险,需要严格控制管理以保证其安全、有效的医疗器械,境内生产企业需要在所在地省(直辖市)药监局进行注册,境外医疗器械生产企业需要委托境内代理人在CFDA进行注册。
第Ⅲ类是具有较高风险,需要采取特别措施严格控制管理以保证其安全、有效的医疗器械,无论境内、境外医疗器械生产企业均需要在CFDA进行注册。
2.注册条件
申报主体
拟从事医疗器械生产且在境内销售的境内生产企业
拟出口医疗器械到中国的境外生产企业
拟在中国大陆销售医疗器械产品的港澳台地区的生产企业
注:境外及港澳台生产企业必须委托在中国境内设立的分支机构或指定的代理人进行注册(备案)申报。
医疗器械注册(备案)类型
中国医疗器械按管理类别分为Ⅰ类、Ⅱ类、Ⅲ类医疗器械,具体产品管理类别的归属,以国家食品药品监督管理部门发布的《医疗器械分类规则》、《医疗器械分类目录》以及不定时发布的分类界定通知为主要依据。
按照医疗器械的类别、注册不同阶段等情况,医疗器械的注册可以分为首次注册、注册变更、延续注册三个类型。
| 产品类别 | 注册类型 | 适用范围 |
| 非体外诊断试剂 (无源医疗器械、有源医疗器械) | 备案 | 无论境外如何管理,境内按第Ⅰ类医疗器械管理的产品 |
| 首次注册 | (1)境外按医疗器械管理的已上市的第Ⅱ、Ⅲ类医疗器械产品首次进入中国市场 (2)境外已上市且不按医疗器械管理而境内需按医疗器械管理的第Ⅱ、Ⅲ类产品首次进入中国市场 | |
| 注册变更 | 登记事项变更: (1)申请人名称; (2)申请人注册地址 (3)生产地址(文字性改变) (4)代理人名称 (5)代理人注册地址 许可事项变更: (1)产品名称 (2)型号、规格; (3)产品性能结构及组成; (4)产品适用范围。 | |
| 延续注册 | (1)到期重新注册 (2)在医疗器械注册证有效期届满6个月前、且有效期届满前12个月内 (3)改变管理类别延续注册 (4)管理类别由高类别调整为低类别的,在有效期内的医疗器械注册证继续有效。如需延续的,申请人应当在医疗器械注册证书有效期届满6个月前、且有效期届满前12个月内,按照改变后的类别到相应的食品药品监督管理部门申请延续注册或办理备案。 (5)管理类别由低类别调整为高类别的,申请人应当按照本办法第六章的规定,在类别调整后6个月内,按照改变后的类别向相应的食品药品监督管理部门申请注册。 | |
| 体外诊断试剂 | 备案 | 无论境外如何管理,境内按第Ⅰ类医疗器械管理的产品 |
| 首次注册 | (1)第Ⅱ、Ⅲ类体外诊断试剂产品首次进入中国市场; (2)已上市销售产品基本反应原理改变; (3)已上市销售产品分析灵敏度指标改变,并具有新的临床诊断意义; 其他影响产品性能的重大改变。 | |
| 注册变更 | 登记事项变更: (1)变更生产企业名称; (2)变更生产企业注册地址; (3)变更注册代理机构; (4)变更代理人。 许可事项变更: (1)变更生产过程中所用抗原、抗体等主要材料; (2)变更检测条件及参考值(或参考范围)等; (3)变更注册产品标准中所设定的项目、指标、试验方法等; (4)变更产品说明书中的内容,如变更或增加包装规格、增加适用机型等; (5)变更产品储存条件和/或产品有效期; (6)增加临床适用范围,如增加临床适应症、增加临床测定用样本类型等; (7)变更生产地址(生产场所的实质性变更); (8)其他可能影响产品安全性、有效性的变更。 | |
| 延续注册 | 在医疗器械注册证书有效期届满后仍需继续生产、销售的产品应在注册证书有效期届满6个月前、且有效期届满前12个月内提出申请。 |
3.注册申请及审批流程